Menu
")
Quand faut-il évoquer une maladie de Charcot ?
La SLA se traduit par l’association de signes et de symptômes témoignant de l’atteinte des motoneurones du cerveau (« motoneurones centraux ») et de ceux de la moelle épinière et du bulbe cérébral (« motoneurones périphériques »). L’importance des manifestations et les zones touchées sont variables d’une personne à l’autre.
La SLA touche uniquement la motricité et respecte les autres fonctions du système nerveux (pas d’atteinte sensitive), tout comme les 5 sens (vision, ouïe, goût, odorat et toucher). Elle n’affecte pas les muscle des yeux, du cœur, de la vessie, de l’intestin ni des organes sexuels.
Le diagnostic doit être évoqué lors de l’apparition d’une faiblesse musculaire et d’une fonte de la masse musculaire localisée, au niveau des membres inférieurs ou supérieurs. Les muscles se rigidifient, engendrant des difficultés dans la dextérité manuelle et des changements dans la démarche. L’apparition d’une dysarthrie (difficultés d’élocution) et d’une dysphagie (difficultés à déglutir) est assez évocatrice.
La paralysie est progressive. Survient ensuite une atteinte respiratoire allant jusqu’à l’insuffisance respiratoire, qui conduit au décès dans les 3 à 5 ans (parfois, mais rarement, 10 ans).
L’existence de cas de maladie de Charcot dans la famille peut aussi éveiller les soupçons.
Comment diagnostiquer une maladie de Charcot ?
Le diagnostic est essentiellement clinique basé sur l’examen neurologique, et l’électromyogramme (EMG).
Il peut être difficile, notamment au début de la maladie où les signes sont discrets et non caractéristiques.
Il n’y a aucun marqueur biologique de la maladie donc aucune utilité à un bilan sanguin.
Le médecin, neurologue habituellement, va rechercher des signes d’atteinte des motoneurones (raideur des muscles, exagération des réflexes…) ou périphériques (crampes, amyotrophie, fasciculations…).
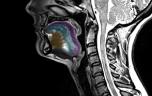
L’électromyogramme (EMG) permet de détecter les anomalies électriques de souffrance des motoneurones et leur étendue. D’autres examens (IRM du cerveau ou médullaire) peuvent être réalisés afin d’éliminer d’autres causes pouvant également expliquer les signes cliniques.
L’étude génétique à la recherche d’une mutation n’est proposée qu’aux formes familiales (au moins deux cas familiaux quel que soit le degré de parenté).
Un établissement lié à un groupe mutualiste innove en matière de bilan de santé : l'utilisation d'un « jumeau numérique » permet ...
Coup de food sur le diabète, c'est votre nouveau rendez-vous culinaire qui bouscule les idées reçues ! Dans cet épisode, une ...
"Les rendez-vous de la santé et de la qualité de vie au travail" de Pourquoi Docteur reçoivent Régis Blugeon, DRH et directeur ...
Dans cette vidéo, le Dr Inès Zaraa, dermatologue à Paris, vous explique comment protéger vos mains au quotidien et éviter les ...
Une plaque rouge qui gratte, une peau sèche qui tiraille, une démangeaison persistante… Et si ce n'était pas juste une irritation ...
« Le terme « eczéma » est souvent utilisé à tort pour désigner n'importe quelle plaque rouge qui gratte. Pourtant, il recouvre ...
"Les rendez-vous de la santé et de la qualité de vie au travail" de Pourquoi Docteur reçoivent Olivier Terral, directeur prévention ...
Retrouvez les conseils précieux et les réponses à vos questions de nos Médecins Experts sur la gestion du diabète pendant le ...
Le Ramadan approche, et, pour de nombreuses personnes atteintes de diabète, c'est une période de questions, de défis, mais ...
Le Dr Julien Poupart, neurologue à l'Hôpital Saint Vincent de Paul - GHICL, Mme Lise Declercq, infirmière au GHICL et Mme ...






































")
")
")






")

